
7: Sequence Alignment
- Page ID
- 93488

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The software program BLAST (Basic Local Alignment Search Tool) uses sequence alignment algorithms to compare a query sequence against a database to identify other known sequences similar to the query sequence. Often, the annotations attached to the already known sequences yield important biological information about the query sequence. Almost all biologists use BLAST, making sequence alignment one of the most important algorithms of bioinformatics.
The sequence under study can be composed of nucleotides (from the nucleic acids DNA or RNA) or amino acids (from proteins). Nucleic acids chain together four different nucleotides: A,C,T,G for DNA and A, C,U,G for RNA; proteins chain together twenty different amino acids. The sequence of a DNA molecule or of a protein is the linear order of nucleotides or amino acids in a specified direction, defined by the chemistry of the molecule. There is no need for us to know the exact details of the chemistry; it is sufficient to know that a protein has distinguishable ends called the N-terminus and the C-terminus, and that the usual convention is to read the amino acid sequence from the N-terminus to the C-terminus. Specification of the direction is more complicated for a DNA molecule than for a protein molecule because of the double helix structure of DNA, and this will be explained in Section 7.1.
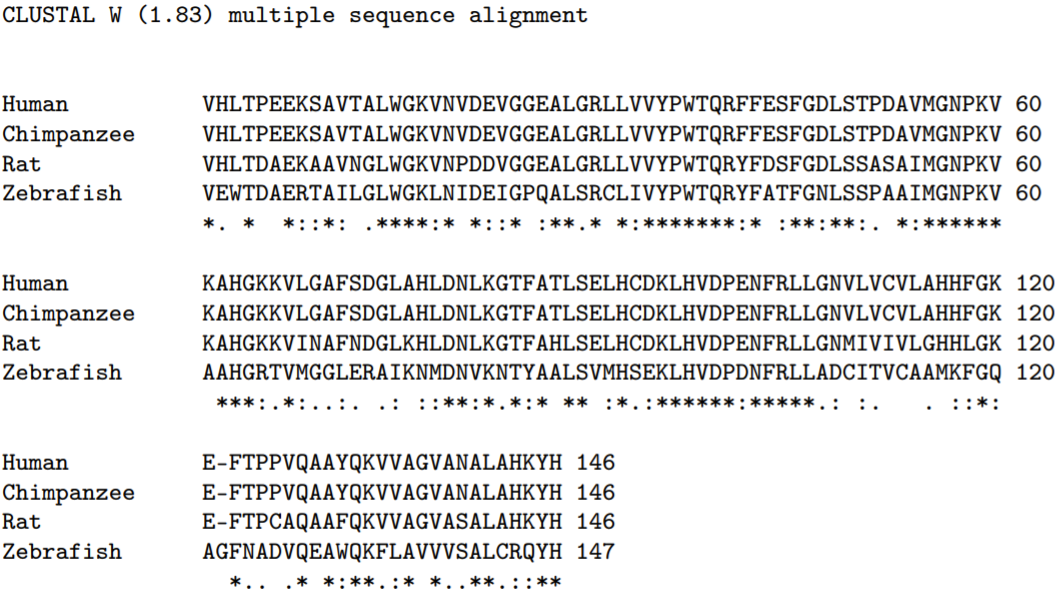
The basic sequence alignment algorithm aligns two or more sequences to highlight their similarity, inserting a small number of gaps into each sequence (usually denoted by dashes) to align wherever possible identical or similar characters. For instance, Fig \(7.1\) presents an alignment using the software tool ClustalW of the hemoglobin beta-chain from a human, a chimpanzee, a rat, and a zebrafish. The human and chimpanzee sequences are identical, a consequence of our very close evolutionary relationship. The rat sequence differs from human/chimpanzee at only 27 out of 146 amino acids; we are all mammals. The zebrafish sequence, though clearly related, diverges significantly. Notice the insertion of a gap in each of the mammal sequences at the zebra fish amino acid position 122 . This permits the subsequent zebrafish sequence to better align with the mammal sequences, and implies either an insertion of a new amino acid in fish, or a deletion of an amino acid in mammals. The insertion or deletion of a character in a sequence is called an indel. Mismatches in sequence, such as that occurring between zebrafish and mammals at amino acid positions 2 and 3 is called a mutation. ClustalW places a "* on the last line to denote exact amino acid matches across all sequences, and \(\mathrm{a}^{\prime}: '\) and ’ \('\) ’ to denote chemically similar amino acids across all sequences (each amino acid has characteristic chemical properties, and amino acids can be grouped according to similar properties). In this chapter, we detail the algorithms used to align sequences.
- 7.2: Brute Force Alignment
- One (bad) approach to sequence alignment is to align the two sequences in all possible ways, score the alignments with an assumed scoring system, and determine the highest scoring alignment. The problem with this brute-force approach is that the number of possible alignments grows exponentially with sequence length; and for sequences of reasonable length, the computation is already impossible.
- 7.3: Dynamic Programming
- Two reasonably sized sequences cannot be aligned by brute force. Luckily, there is another algorithm borrowed from computer science, dynamic programming, that makes use of a dynamic matrix.
- 7.4: Gaps
- Empirical evidence suggests that gaps cluster, in both nucleotide and protein sequences. Clustering is usually modeled by different penalties for gap opening.
- 7.5: Local Alignments
- We have so far discussed how to align two sequences over their entire length, called a global alignment. Often, however, it is more useful to align two sequences over only part of their lengths, called a local alignment. In bioinformatics, the algorithm for global alignment is called "Needleman-Wunsch," and that for local alignment "Smith-Waterman."
- 7.6: Software
- If you have in hand two or more sequences that you would like to align, there is a choice of software tools available.